半导体异质结因其架构构造的特点,具有量子效应、迁移率大等特点,具有奇异的二度空间特性,在发光组件、激光二极管、异质结构双极晶体管等领域有广泛应用。我们以二维MoS
我们首先简单讲一下算法原理,要搭建异质结,为了减少晶格适配率,一般要对结构A和B进行超胞操作。假设结构A超胞的面内晶格常数分别为
显然要使A和B要构成超晶格,必须满足三个条件:(1)
DO m1=0, N
DO n1=0, N
DO l1=0, N
DO k1=0, N
DO m2=0, N
DO n2=0, N
DO l2=0, N
DO k2=0, N
IF((A1-A2) < eps AND (B1-B2) < eps AND (gamma1-gamma2) <eps) THEN
WRITE('This is a heterojunction')
ENDIF
END DO
END DO
END DO
END DO
END DO
END DO
END DO
END DO第一步:准备好黑磷和MoS2原胞结构文件POSCAR1和POSCAR2,当然也可以是其它文件名称,这时候vaspkit会要求你输入文件名;
第二步:运行vaspkit,依次输入以下命令:
------------>>
8
============================ Model Options ======================
803) Build Surface with Specified Miller Index
804) Build Heterostructure from Two Specified Slabs
0) Quit
9) Back
------------>>
804
Input the Mismatch Tolerance (e.g. 0.5 means < 0.5%)
------------>>
0.8 (0.80代表晶格适配率<0.8%)
Input the Interlayer Space and Vacuum Thickness (e.g., 3.0 10):
------------>>
3.0 12(3.0表示搭建异质结时两层Slab之间的距离为3埃,真空层厚度设置为12埃)经过几分钟的计算, 一共得到35个构型满足晶格晶格适配率小于0.5%的条件,vaspkit会一次性生成所有满足限定条件的结构。我们以HETERO_5_46.0_192.vasp文件来说明生成文件命名规则:5代表第5个构型,46.0表示异质结面内晶格基矢夹角为46°,一共有192个原子。更具体的结构信息可通过HETERO_LIST文件查看,结构信息格式如下所示:
Configuration: 1
Mismatch (%): 0.568
Lattice Angle (Degree): 49.006
Lattice Constant a and b (Angstrom): 30.263 19.852
Total atoms: 276
Total atoms of A and B Structures: 120 156
Transformation matrix for A Structure:
6 5 0
6 0 0
0 0 1
Transformation matrix for B Structure:
10 9 0
2 7 0
0 0 1
<<--------------------------------------------------------------------->>
Configuration: 2
Mismatch (%): 0.522
Lattice Angle (Degree): 8.529
Lattice Constant a and b (Angstrom): 33.484 30.438
Total atoms: 91
Total atoms of A and B Structures: 40 51
Transformation matrix for A Structure:
3 7 0
4 6 0
0 0 1
Transformation matrix for B Structure:
7 12 0
5 11 0
0 0 1我们随机挑出3个结构如下图所示:
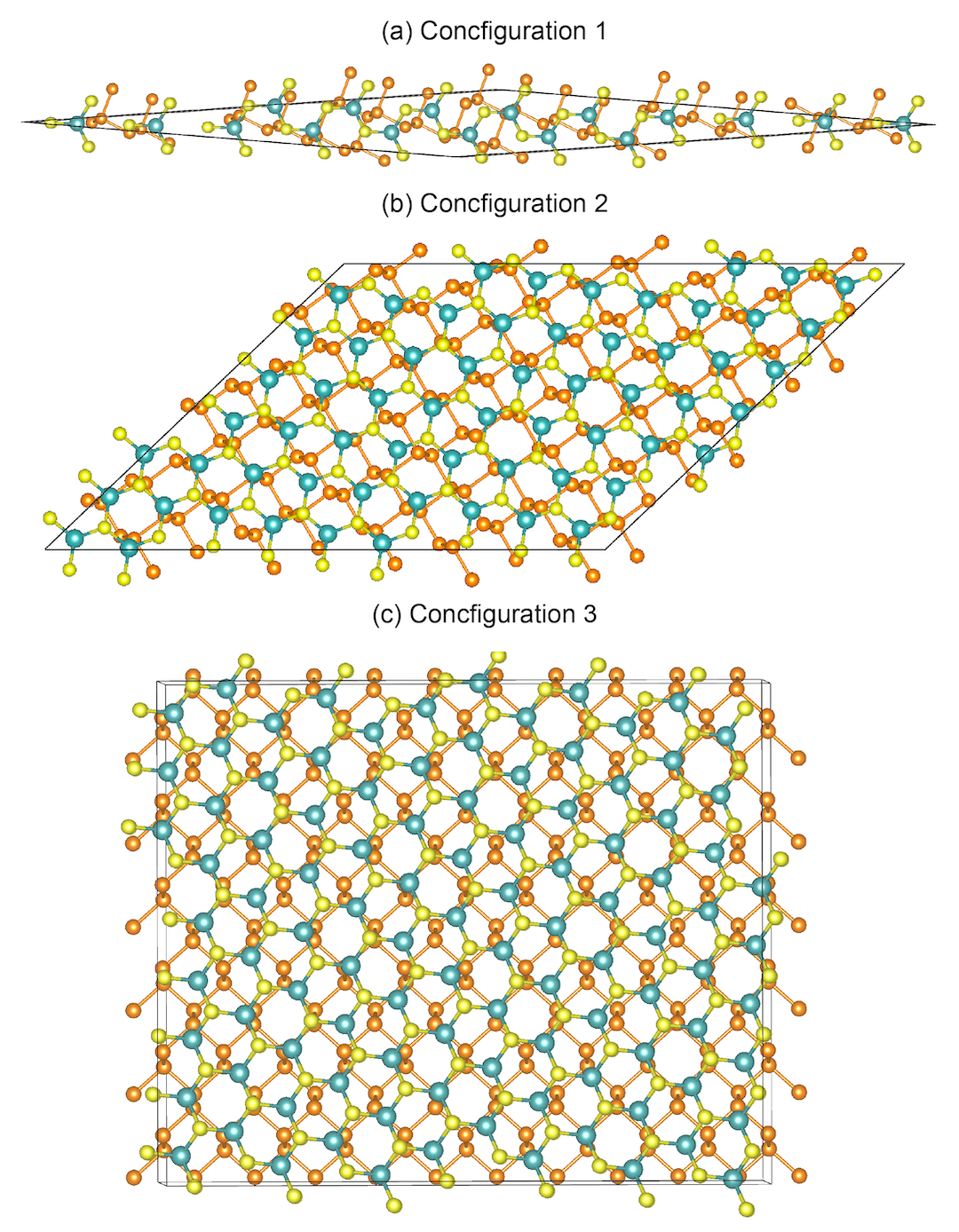
如何想要筛选特定角度的异质结(仅支持1.3.0及以上版本),例如想得到具有正交格子的异质结构,可在~/.vaspkit中设置
MAX_ATOM_NUMBER 1000 最大原子数不超过1000
MIN_LATTICE_ANGLE 89 最小ab晶格夹角为89°
MAX_LATTICE_ANGLE 91 最大ab晶格夹角为91°如果您使用VASPKIT,请记得引用哦!
V. Wang, N. Xu, J.-C. Liu, G. Tang, W.-T. Geng, VASPKIT: A User-Friendly Interface Facilitating High-Throughput Computing and Analysis Using VASP Code, Computer Physics Communications 267, 108033, (2021), https://doi.org/10.1016/j.cpc.2021.108033
欢迎关注VASPKIT公众号。

「感觉有帮助?一键投喂 牛奶/咖啡/冰阔乐!」
 VASPKIT
VASPKIT
(๑>ڡ<)☆哇~太棒了!
使用微信扫描二维码完成支付

你好,有没有办法定义晶格失配度呢,比如我想要1%-2%。我试了原来那样去定义,但是发现会生成大于5000个文件导致运行失败,那些晶格错配度太小的,原子数太多,根本算不动。还有,如果我想要原子位置处于顶为,桥位和中间位又该怎么操作呢
谢谢建议,后续版本会增加最多结构数目。
MS软件里面,可以自己定义晶格失配,关于软件建模这些,我自己的意见还是手动操作比较好些。
你好 这最后输入这个~/.vaspkit这步操作是在哪里输入啊 本人萌新 不知咋操作
请问您会怎么操作了么
vi ~/.vaspkit
相同材料相同的晶面,提供不同夹角的slab生成的结构不一样怎么解决?例如输入AlN(0001)面,分别提供夹角为120度和90度的两个slab,但都是AlN(0001)面。在与另一个slab生成异质结构的时候,生成完成不同的两种结果。这种情况怎么处理?