VASPKIT.1.3.1版本已实现基于半经验玻尔兹曼输运理论计算输运性质,目前可输出electronic conductivity, Seebeck coefficient, power factor 和electronic thermal conductivity等输运性质,后期将逐步改进输运性质计算模块。我们简述一下实现原理。根据玻耳兹曼理论, 电子的群速度可以表示为:
式 中,
上式中,
式中
其中电导系数可表示为
上式中
第一步:准备POSCAR和INCAR文件
准备POSCAR文件
Ga1 As1
1.0
4.0659928322 0.0000000000 0.0000000000
2.0329964161 3.5212530843 0.0000000000
2.0329964161 1.1737510281 3.3198692456
Ga As
1 1
Direct
0.000000000 0.000000000 0.000000000
0.750000000 0.750000000 0.750000000INCAR文件为
Global Parameters
ISTART = 1 (Read existing wavefunction, if there)
ISPIN = 1 (Non-Spin polarised DFT)
LREAL = .FALSE. (Projection operators: automatic)
LWAVE = .TRUE. (Write WAVECAR or not)
LCHARG = .TRUE. (Write CHGCAR or not)
ADDGRID= .TRUE. (Increase grid, helps GGA convergence)
Static Calculation
ISMEAR = 0 (gaussian smearing method)
SIGMA = 0.05 (please check the width of the smearing)
LORBIT = 11 (PAW radii for projected DOS)
NEDOS = 2001 (DOSCAR points)
NELM = 60 (Max electronic SCF steps)
EDIFF = 1E-08 (SCF energy convergence, in eV)第二步:调用VASPKIT-681命令产生用于输运性质计算的KPOINTS文件,运行过程如下
-->> (01) Reading Structural Parameters from POSCAR File...
+-------------------------- Warm Tips --------------------------+
* Accuracy Levels: (1) Medium: 0.02~0.01;
(2) Fine: < 0.01;
* 0.01 is Generally Precise Enough!
+---------------------------------------------------------------+
Input the K-Spacing Value (in Unit of 2*PI/Angstrom):
------------>>
0.005
Reciprocal Lattice Vectors (in Units of 1/Angstrom):
1.5453016192 -0.8921803058 -0.6308667443
0.0000000000 1.7843606116 -0.6308667443
0.0000000000 0.0000000000 1.8926002328
Reciprocal Lattice Constants: 1.8926 1.8926 1.8926
Real-Space Lattice Constants: 4.0660 4.0660 4.0660
Size of K-Mesh: 60 60 60
+---------------------------------------------------------------+
-->> (02) Written KPOINTS File!注意输运性质计算需要极密的K点网格,推荐
第三步:准备好VASP四个输入文件后执行VASP计算
第四步:准备用于计算输运性质的INPUT.in文件(注意VPKIT.in将被统一重命名为INPUT.in)
1 # 1 for gradient method with constant relaxation time approximation
3D # 2D for slab, 3D for bulk
-2.0 2.0 # Minimum and maximum values of chemical potential with respect to Fermi energy
1000 # Number of intervals between the minimum and maximum chemical potential values
300.0 # Temperature (float type, in units of K)
1.0 # Relaxation time (float type, in units of s), Set 1.0 if you don't know the exact relaxation time
0.0 # Desired gap value in scissor correction (float type, in units of V), Zero means make no correction, available only non-spin-polarization calculation
1 # Finite difference method (integer type)
第一行目前只能设置为1,采用常数弛豫时间计算性质。最后一行的数字1表示用于计算群速度的差分方法:设置为1则采用
第五步:调用VASPKIT-682命令计算输运性质,运行过程如下
------------>>
682
-->> (01) Reading INPUT.in File...
-->> (02) Reading Structural Parameters from POSCAR File...
-->> (03) Reading Input Parameters From INCAR File...
+---------------------------------------------------------------+
| >>> The Fermi Energy will be set to zero eV <<< |
+---------------------------------------------------------------+
-->> (04) Reading Fermi-Energy from DOSCAR File...
-->> (05) Reading Energy-Levels From EIGENVAL File...
+-------------------------- Summary ----------------------------+
Chemical Potential Range (eV): -2.00 2.00
Number of Intervals: 1000
Temperature (K): 300.00
Relaxation Time (s): 0.10E+01
+---------------------------------------------------------------+
-->> (06) Calculating Boltzmann Transport Properties...
Percentage complete: 25.0%
Percentage complete: 50.0%
Percentage complete: 75.0%
Percentage complete: 100.0%
-->> (07) Written ELECTRONIC_CONDUCTIVITY.dat File!
-->> (08) Written SEEBECK_COEFFICIENT.dat File!
-->> (09) Written POWER_FACTOR.dat File!
-->> (10) Written ELECTRONIC_THERMAL_CONDUCTIVITY.dat File!
-->> (11) Written CARRIER_CONCENTRATION.dat File!
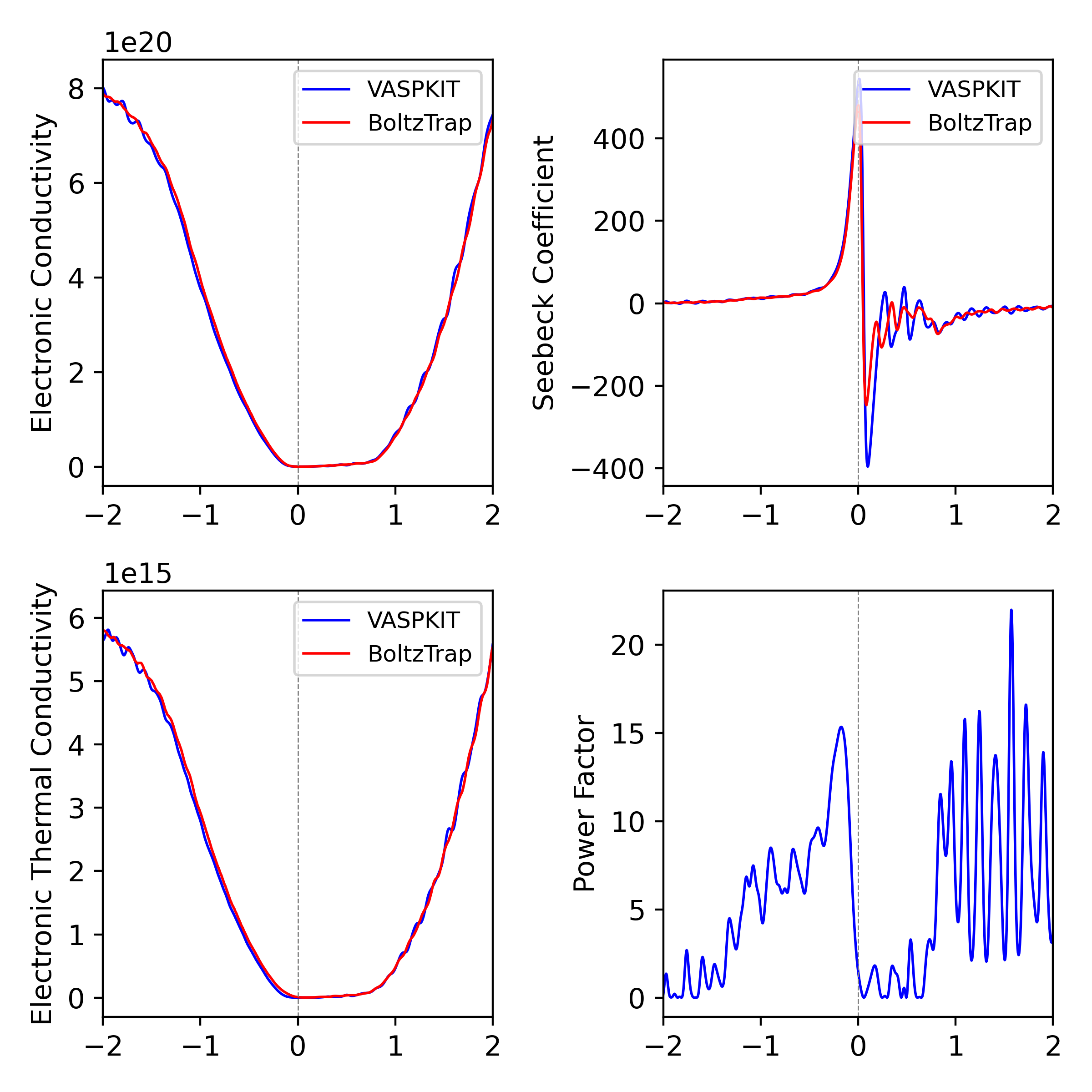
我们的计算结果与Boltztrap程序计算结果非常接近,可进一步增加K点得到平滑的曲线。目前该功能仍需广泛测试,建议找测试体系与Boltztrap结果比较。
如果您使用VASPKIT,请记得引用哦!
V. Wang, N. Xu, J.-C. Liu, G. Tang, W.-T. Geng, VASPKIT: A User-Friendly Interface Facilitating High-Throughput Computing and Analysis Using VASP Code, Computer Physics Communications 267, 108033, (2021), https://doi.org/10.1016/j.cpc.2021.108033
欢迎关注VASPKIT公众号。

「感觉有帮助?一键投喂 牛奶/咖啡/冰阔乐!」
 VASPKIT
VASPKIT
(๑>ڡ<)☆哇~太棒了!
使用微信扫描二维码完成支付

计算输运性质的INPUT.in文件是怎么导入的
vaspkit/examples/Boltzmann_transport例子提供一个INPUT.in模板,在此基础上修改。
这个好像是自己输入的,我自己照着他的写了一遍,出来的图像是一样的,用在其他材料的计算上面应该只需要调节一下电压范围和温度就够了
您好,请问算出来的结果是-2到2eV的数据,怎么得到具体值
请问这个问题你解决了吗?哪一个值为计算结果
您好,您是怎么取值的
电导率不是一个固定值吗,单位%IACS
请问这个结果的图是怎么画的?
该图使用Matplotlib得到,origin也可以啊,简单的二维曲线图。
这个得到的结果应该是弛豫时间设为1s的电导率、热导率,所以才那么大,这个和用boltztrap得到的没有啥本质区别啊,还是得乘一个fs量级的弛豫时间才行
你好,可以留一下联系方式吗?我算出的这个结果这么大到底应该怎么处理
你好,请问你的问题解决了吗?如何才能得到结构的电导率?
主要是他这个假设的弛豫时间是1,这个跟实际的电子弛豫时间差别是很大的,还是得自己另外计算个弛豫时间
弛豫时间怎么算啊?请问
不应该是某一温度下的具体值?
调用682和681命令显示参数错误是怎么回事呀
xy方向的电导率怎么计算
请问计算二维材料时,无论取多大K点都提示要提高精度,是否是需要在真空层方向增加密度
请问你解决了吗,我也遇到了
关于这些性质,有相关论文的分析嘛,想学习一下~
您好,可以加个联系方式讨论学习计算电导吗 qq:2858212544
你好,请问你的问题解决了吗
vaspkit计算bolztrap时化学势怎么选取呀?我得到的极大值的极小值并不在0点附近,请问是什么原因?
您好,可以加个联系方式讨论学习计算电导吗 qq:2858212544
您好,681生成kpoints第四列是权重吗
是权重,为了减少计算量,只需要在不可约布里渊区产生K点。
您好,请问怎么计算自旋电导率?
我目的是想计算自旋电荷转化比,然后思路是先计算x方向的电导率,再把y方向的电导率分成自旋向上和自旋向下的,再作差,把作差的结果除以x方向的电导率,得到自旋电荷转化比。公式:(σxy up -σxy down)/σxx
您好,请问计算出来的载流子浓度上负号代表载流子为电子,正号表示载流子为空穴对吗
这里的横坐标表示的是什么呀
你好,有答案了吗
是那个能量,input文件里那个化学势与费米能的差
电子群速度是怎么算的
请问这个结果有没有矫正过费米能级?
请问我是按照教程得到的KPOINTS,但是vaspkit得到结果时报错 Please increase k-spacing (k>4) when generate KPOINTS file,我应该怎么做
我也遇到了这个问题,请问您解决了吗
这个可以计算得到载流子浓度吗?
有载流子浓度输出
请问这个直接得到的是300K室温下的电导率吗
温度是你自己设置的
画出来的三条线表示什么啊 他们没有重合在一起
你好,请问INPUT.in文件倒数第二行是写pbe与实验的差值吗 但是好像单位不一样,是要转换为V吗
请问可以计算金属体系的电导率吗
请问在用vaspkit自带的玻尔兹曼输运程序的时候能否输出非对角线的电导项呢,比如霍尔电导项sigma_xy?
电导率的单位是什么呢,为啥是一个范围?
请问这个ISPIN=2,可以吗,因为是磁性体系
请问计算这个电子运输性质很吃计算机内存吗?我的96原子体系超算内存用到2T都存在内存溢出现象。
相较于静态自洽电荷计算的INCAR设置只多了一个NEDOS=2001。