在材料的线性形变范围内(应变较小的情况下),体系的应力与应变满足胡克定律,
施加应变后体系的应变能
这里
因此,采用第一性原理计算弹性常数有两种方法,第一种方法是应力-应变方法,即通过给结构施加不同应变,分别计算出所对应的应力大小,然后利用公式(1)拟合得到一次项系数,从而得到
这里我们以金刚石结构为例讲解如何采用能量-应变函数关系计算弹性常数,详见vaspkit/examples/elastic/diamond_3D。对于金刚石立方结构,一共有3个独立弹性常数,C11,C12和C44。
立方晶系的弹性能可以表示
设定
设定
再设定
联立以上三个方程组,即可得到三个独立弹性常数。
施加形变后的晶格基矢可通过下式得到 [2]:
其中
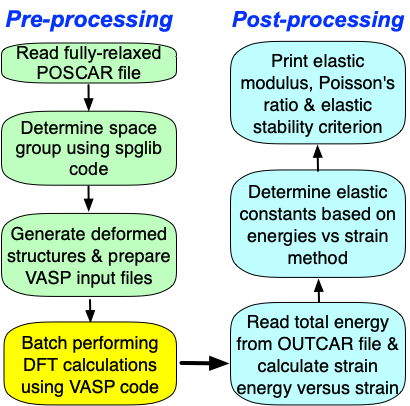
VASPKIT和VASP计算晶体的弹性常数具体计算步骤分为:
1,准备优化彻底的POSCAR文件,注意通常采用标准的惯用原胞计算弹性常数,如果不确信POSCAR文件中是否是标准的惯用原胞,可以用vaspkit-603/604生成标准结构;
2,运行vaspkit 选择102生成KPOINTS,由于计算弹性常数对K-mesh要求很高,因此对于半导体(金属体)体系,生成K点的精度应不小于
3,INCAR参考设置如下;
Global Parameters
ISTART = 0
LREAL = F
PREC = High (截断能设置默认值1.5-2倍)
LWAVE = F
LCHARG = F
ADDGRID= .TRUE.
Electronic Relaxation
ISMEAR = 0
SIGMA = 0.05
NELM = 40
NELMIN = 4
EDIFF = 1E-08
Ionic Relaxation
NELMIN = 6
NSW = 100
IBRION = 2
ISIF = 2 (切记选择2,如果选择3会把施加应变后原胞重新优化成平衡原胞)
EDIFFG = -1E-02 4,准备VPKIT.in文件并设置第一行为1(预处理);运行vaspkit并选择201, 将生成用于计算弹性常数文件;
1 ! 设置1将产生计算弹性常数的输入文件,2则计算弹性常数
3D ! 2D为二维体系,3D为三维体系
7 ! 7个应变
-0.015 -0.010 -0.005 0.000 0.005 0.010 0.015 ! 应变变化范围运行vaspkit后输出一下信息
------------>>
201
-->> (01) Reading VPKIT.in File...
+-------------------------- Warm Tips --------------------------+
See an example in vaspkit/examples/elastic/diamond_3D,
Require the fully-relaxed and standardized Convertional cell.
+---------------------------------------------------------------+
-->> (02) Reading Structural Parameters from POSCAR File...
-> C44 folder created successfully!
-> strain_-0.015 folder created successfully!
-> strain_-0.010 folder created successfully!
-> strain_-0.005 folder created successfully!
-> strain_0.000 folder created successfully!
-> strain_+0.005 folder created successfully!
-> strain_+0.010 folder created successfully!
-> strain_+0.015 folder created successfully!
-> C11_C12_I folder created successfully!
-> strain_-0.015 folder created successfully!
-> strain_-0.010 folder created successfully!
-> strain_-0.005 folder created successfully!
-> strain_0.000 folder created successfully!
-> strain_+0.005 folder created successfully!
-> strain_+0.010 folder created successfully!
-> strain_+0.015 folder created successfully!
-> C11_C12_II folder created successfully!
-> strain_-0.015 folder created successfully!
-> strain_-0.010 folder created successfully!
-> strain_-0.005 folder created successfully!
-> strain_0.000 folder created successfully!
-> strain_+0.005 folder created successfully!
-> strain_+0.010 folder created successfully!
-> strain_+0.015 folder created successfully!5,批量提交vasp作业;
6,再次修改VPKIT.in文件中第一行为2(后处理),然后再次运行vaspkit并选择201,得到以下结果;
------------>>
201
-->> (01) Reading VPKIT.in File...
+-------------------------- Warm Tips --------------------------+
See an example in vaspkit/examples/elastic/diamond_3D,
Require the fully-relaxed and standardized Convertional cell.
+---------------------------------------------------------------+
-->> (02) Reading Structural Parameters from POSCAR File...
-->> (03) Calculating the fitting coefficients of energy vs strain.
-->> Current directory: Fitting Precision
C44: 0.817E-09
C11_C12_I: 0.814E-08
C11_C12_II: 0.135E-07
+-------------------------- Summary ----------------------------+
Based on the Strain versus Energy method.
Crystal Class: m-3m
Space Group: Fd-3m
Crystal System: Cubic system
Including Point group classes: 23, 2/m-3, 432, -43m, 4/m-32/m
There are 3 independent elastic constants
C11 C12 C12 0 0 0
C12 C11 C12 0 0 0
C12 C12 C11 0 0 0
0 0 0 C44 0 0
0 0 0 0 C44 0
0 0 0 0 0 C44
Stiffness Tensor C_ij (in GPa):
1050.640 126.640 126.640 0.000 0.000 0.000
126.640 1050.640 126.640 0.000 0.000 0.000
126.640 126.640 1050.640 0.000 0.000 0.000
0.000 0.000 0.000 559.861 0.000 0.000
0.000 0.000 0.000 0.000 559.861 0.000
0.000 0.000 0.000 0.000 0.000 559.861
Compliance Tensor S_ij (in GPa^{-1}):
0.000977 -0.000105 -0.000105 0.000000 0.000000 0.000000
-0.000105 0.000977 -0.000105 0.000000 0.000000 0.000000
-0.000105 -0.000105 0.000977 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.001786 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.001786 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.001786
Elastic stability criteria as discussed in PRB 90, 224104 (2014):
Criteria (i) C11 - C12 > 0 meeted.
Criteria (ii) C11 + 2C12 > 0 meeted.
Criteria (iii) C44 > 0 meeted.
Average mechanical properties for polycrystalline:
+---------------------------------------------------------------+
| Scheme | Voigt | Reuss | Hill |
+---------------------------------------------------------------+
| Bulk modulus K (GPa) | 434.640 | 434.640 | 434.640 |
| Shear modulus G (GPa) | 520.716 | 516.130 | 518.423 |
| Young's modulus E (GPa) | 1116.342 | 1109.298 | 1112.824 |
| P-wave modulus (GPa) | 1128.929 | 1122.814 | 1125.871 |
| Poisson's ratio v | 0.072 | 0.075 | 0.073 |
| Bulk/Shear ratio | 0.835 | 0.842 | 0.838 |
+---------------------------------------------------------------+
Pugh Ratio: 1.193
Cauchy Pressure (GPa): -433.220
Universal Elastic Anisotropy: 0.044
Chung–Buessem Anisotropy: 0.004
Isotropic Poisson’s Ratio: 0.073
Longitudinal wave velocity (in m/s): 17945.173
Transverse wave velocity (in m/s): 12177.146
Average wave velocity (in m/s): 13280.911
Debye temperature (in K): 2212.889
References:
[1] Voigt W, Lehrbuch der Kristallphysik (1928)
[2] Reuss A, Z. Angew. Math. Mech. 9 49–58 (1929)
[3] Hill R, Proc. Phys. Soc. A 65 349–54 (1952)
[4] Debye temperature J. Phys. Chem. Solids 24, 909-917 (1963)
[5] Elastic wave velocities calculated using Navier's equation金刚石弹性常数实验值分别为:
如果在计算弹性常数时希望强制固定某个体系的空间群,可在SYMMETRY.in文件第二行设置空间群,这样晶系将通过指定空间群判断并计算相应的独立弹性常数,这对于掺杂或者合金体系比较有用。
参考文献:
1 武松,利用 VASP 计算不同晶系晶体弹性常数
[2] 侯柱锋,采用VASP如何计算晶体的弹性常数
[3] McSkimin H J and Andreatch P 1972 J. Appl. Phys. 43 2944
如果您使用VASPKIT,请记得引用哦!
V. Wang, N. Xu, J.-C. Liu, G. Tang, W.-T. Geng, VASPKIT: A User-Friendly Interface Facilitating High-Throughput Computing and Analysis Using VASP Code, Computer Physics Communications 267, 108033, (2021), https://doi.org/10.1016/j.cpc.2021.108033
欢迎关注VASPKIT公众号。

「感觉有帮助?一键投喂 牛奶/咖啡/冰阔乐!」
 VASPKIT
VASPKIT
(๑>ڡ<)☆哇~太棒了!
使用微信扫描二维码完成支付

你好,我想请问一下怎么批量提交vasp作业呢
用bash循环遍历每个任务文件夹,可参考vaspkit/examples/diamond_3D中的submit_batch_vasp_jobs.sh批量提交作业脚本。
2维材料只得到了3*3的刚度矩阵咋处理呢
保存到ELASTIC_TENSOR_2D.in文件中,注意第一行是注释行必须有,然后运行vaspkit-204。
######################################################
Read Elastic Constants fo 2D Material from the ELASTIC_TENSOR_2D.in file if it exists.
104.4790 21.5850 0.0000
21.5850 34.0190 0.0000
0.0000 0.0000 27.4020
######################################################
您好为何我进行到将vpkit.in修改为2时 系统会提示错误警告说-0.015时候的outcar不存在啊求解答。
我的也是 你解决了吗
解决了吗请问
老哥,你解决了么
请问您解决了吗,我也遇到这个问题了
你好,请问你解决了吗这个问题(!)[zface_4.png] (!)[zface_4.png]
你好,请问你解决了吗这个问题(!)[zface_4.png]
您好,请问解决了吗
用vaspkit对弹性矩阵后处理之后,会得到两个硬度值,请问第二个硬度值所引用的参考文献 Tian Y-J, Int. J. Refract. Hard Met. 33, 93–106 (2012),具体是用的哪个微观模型计算的?
请问2/3里步骤准备的KPOINTS和INCAR是给后续批量计算准备的还是在讲第1步里“准备优化彻底的POSCAR”的方法呢?有点懵
你好,我想问一下我这里最后一步报错Error: The calculation in is abnormal end.怎么办?@VASPKIT
每一个都输出了reached required accuracy - stopping structural energy minimisation
为什么我的是出来很多文件夹C11 C11_C12_C22等很多文件,我看这里面只出现了三个独立的弹性模量文件,是我哪里操作的有问题吗
请问一下看最后结果时WARNING! The Unstrained Structure Has Non-Zero Elastic-Energy.|
| I Strongly Suggest You Reperform Structure Optimization Again.报这个错误,一般是什么原因,怎么处理
请问你这个问题解决了吗,我也遇到了一样的问题,可以请教一下吗
没解决,我结构有问题
请问你解决了吗?我现在也遇到这样的问题了
您好,我在计算MoC的弹性常数时先试用了vaspkit603生成了惯用晶胞,但是里面只有一个Mo和一个C,而后在后处理的时候vaspkit识别了C66分量,但是按理说六方结构是不会有C66分量的
老师你好Current directory: Fitting Precision,这个值在多少的时候,数据比较可信呢?
请问这里输的应变是单轴双轴还是静水的应变。