态密度很多分析和能带的分析结果可以一一对应,很多术语也和能带分析相通。但是因为它更直观,因此在结果讨论中用得比能带分析更广泛一些。在电子能级为准连续分布的情况下,单位能量间隔内的电子态数目。即能量介于LORBIT=10计算的就是LDOS,也就是每个原子的局域态密度 (local DOS),是分解到每个原子上面的s,p,d;LORBIT=11,计算的就是PDOS,投影态密度(projected DOS)或分波态密度(partial DOS),不仅分解到每个原子的s,p,d,而且还进行px,py,pz分解。
vaspkit最近优化了DOS的提取功能。相对于P4VASP提取DOS信息,vaspkit有以下几个优点:①不需要将体积庞大的vasprun.xml拖回本地;②支持f轨道的提取,P4VASP提取f轨道时存在bug;③生成的dat文件格式友好,而P4VASP导出的dat是以空格为分隔符,无法直接用Origin绘图,并且没有DOS线的图例。
以官网的CO在Ni表面的吸附模型和Ni 100 surface为例提取 PDOS。
CO吸附的例子中设置了LORBIT=11投影了轨道,关闭了自旋极化。
在/vaspkit.0.72/examples/LDOS_PDOS/Partial_DOS_of_CO_on_Ni_111_surface目录下启动vaspkit,输入命令115选择子功能The Sum of Projected DOS for Selected Atoms and orbitals。我们需要提取的是O的s,p轨道,C的s,p轨道以及表面的dxy和d3z2-r2(dz2)轨道。首先提示你选择元素(累加),第一次输入元素O,回车后提示你输入提取的轨道名(累加),第一次输入轨道s。回车后重复以上两个操作。如果想结束输入,在元素选择行直接按回车键结束输入。
Input the Symbol and/or Number of Atoms to Sum [Total-atom: 7]
(Free Format is OK, e.g., C Fe H 1-4 7 8 24),Press "Enter" if you want to end entry!
------------>>
O
Input the Orbitals to Sum
Which orbital? s py pz px dxy dyz dz2 dxz dx2 f-3 ~ f3, "all" for summing ALL.
s本次的输入内容如下:
O - s - O - p - C - s - C - p - Ni - dxy - Ni - dz2 - 'Enter'在目录下会生成一个PDOS_USER.dat,内容如下:
#Energy O_s O_p C_s C_p Ni_dxy Ni_dz2
-27.10266 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000
-26.92966 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000
-26.75566 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000
-26.58266 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000第一行是列名,也就是轨道的名称,2-301行为PDOS的数据点。拖入Origin可以得到PDOS图,官网的图也放在下面供参考。我们也提供了一个DOS_plot.py脚本,在python2.7环境下能够直接绘制PDOS图。VASP5.4.4计算的O的s轨道的PDOS会比官网例子低一点,此现象已经用P4VASP验证无误。
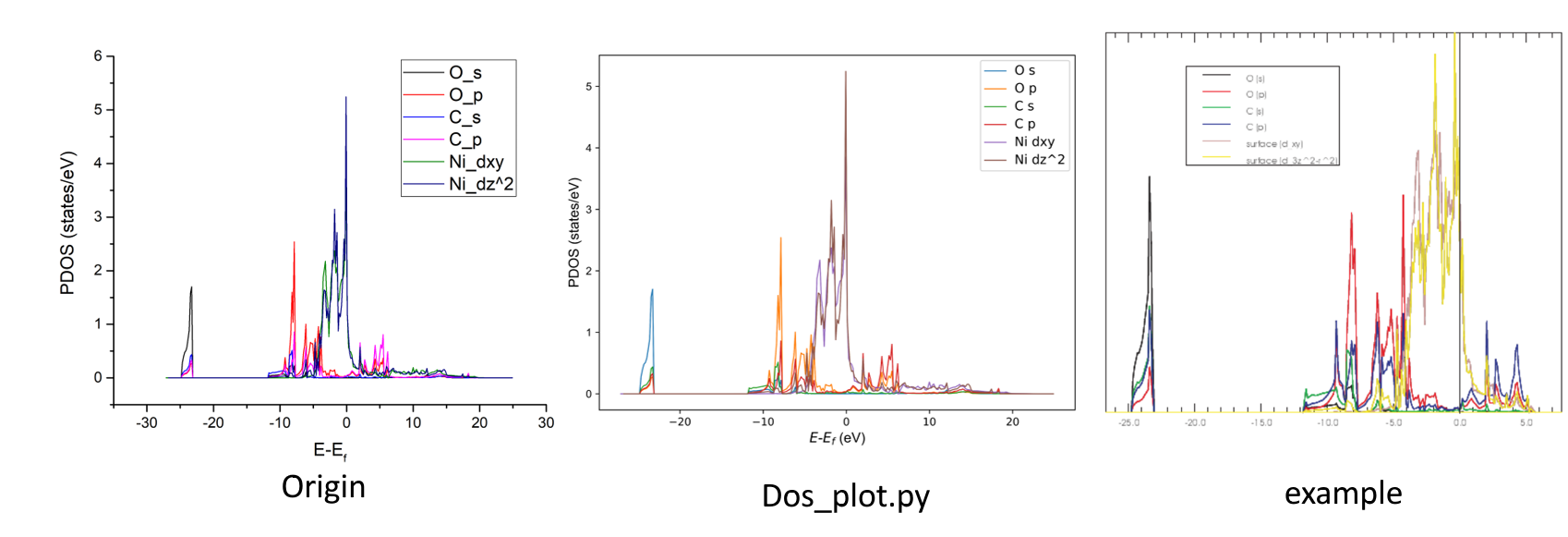
元素行接受自由格式输入,你可以输入以下内容1-3 4 Ni表示选择元素1,2,3,4和Ni元素的PDOS进行累加。轨道行只支持标准输入,如果使用了LORBIT=10不投影轨道,你只能选择 s p d f中的一个或多个,如果使用了LORBIT=11投影了轨道,你可以从s p d f和s py pz px dxy dyz dz2 dxz dx2 f-3 f-2 f-1 f0 f1 f2 f3中选择一个或多个输入。轨道行还支持输入all计算所有轨道的DOS之和。
比如元素行输入C O ,对应的轨道选择s px,那么提取的就是所有C和O元素的s轨道和px轨道的PDOS之和。 PDOS_USER.dat中的轨道名 为#Energy C&O_s&px,显而易懂。
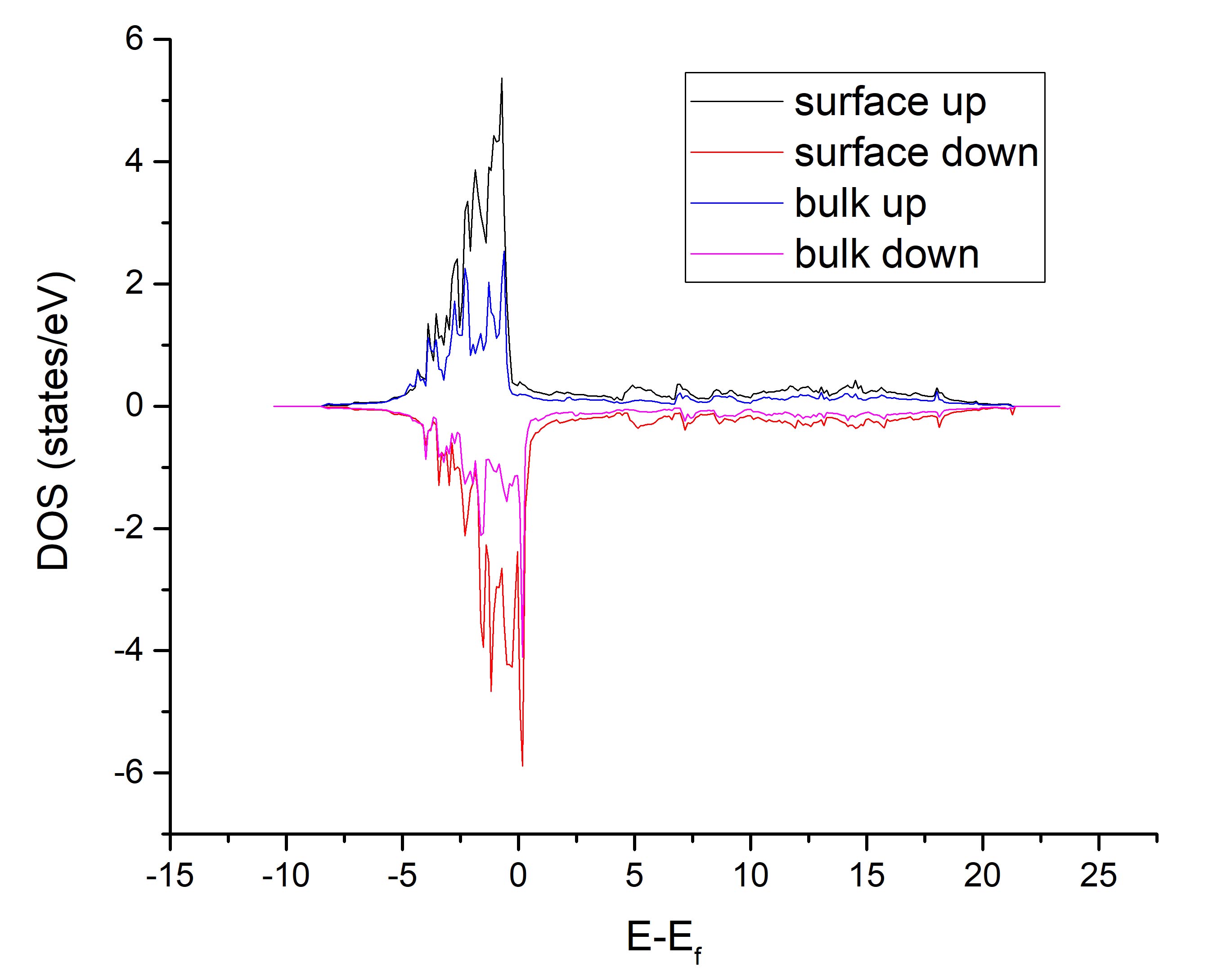
接下来以纯的Ni表面为例。vaspkit/examples/LDOS_PDOS/Ni_100_surface_DOS
本例设置了LORBIT=10,开启了自旋极化。
本次的输入内容如下:
1 5 - all - 3 - all - 'Enter'提取的是上下Ni表面的总态密度和中间bulk层的总态密度。
PDOS_USER.dat的内容如下:
#Energy up_1&5_all dw_1&5_all up_3_all dw_3_all
-10.54087 0.00000 0.00000 0.00000 0.00000
-10.42787 0.00000 0.00000 0.00000 0.00000
-10.31487 0.00000 0.00000 0.00000 0.00000第二列为上下表面的spin up的总态密度,第三列为上下表面的spin down的总态密度。拖进Origin,可以轻松做出 DOS图。
如果您使用VASPKIT,请记得引用哦!
V. Wang, N. Xu, J.-C. Liu, G. Tang, W.-T. Geng, VASPKIT: A User-Friendly Interface Facilitating High-Throughput Computing and Analysis Using VASP Code, Computer Physics Communications 267, 108033, (2021), https://doi.org/10.1016/j.cpc.2021.108033
欢迎关注VASPKIT公众号。

「感觉有帮助?一键投喂 牛奶/咖啡/冰阔乐!」
 VASPKIT
VASPKIT
(๑>ڡ<)☆哇~太棒了!
使用微信扫描二维码完成支付

DOS_plot.py脚本在哪下载呢
请问,如果设置LORBIT=11时,产生每个原子的px,py,pz态密度,这时的px,py,pz相加是否等于LORBIT=10时产生的p轨道的态密度。
找几个例子验证一下。即使有差别也会很小。
你好,请问自旋极化在结构优化的时候打开嘛
好像有磁性的原子要开,没有的不开
[...]http://vaspkit.cn/index.php/33.html(vaspkit处理态密度)[...]
O - s - O - p - C - s - C - p - Ni - dxy - Ni - dz2 - 'Enter'。中间的 - 可以理解为“Enter”(!)[zface_7.png]
如何提取总体系的分波态密度啊,请问,帖子里面的我看见是分别提取不同原子的分波态密度
是不是如果原子的轨道是空的,如dx2-y2,那么输出的数据会失败??会出现这样的提示, Selected orbitals are not in the output!