能带计算的KPOINTS与普通计算的KPOINTS不一样,通常需要第一布里渊区内的一条或几条高对称点路径来计算能带性质。传统的做法是通过SeeK-Path网站或者Material Studio软件获得晶体倒易空间第一布里渊区内的高对称点,再通过脚本插值生成高对称点路径上的K点,得到满足要求的KPOINTS。好消息是新版的vaspkit集成了与SeeK-path一致的算法分析晶体的高对称点,可以方便地生成PBE泛函和HSE06杂化泛函所需的KPOINTS。
在vaspkit/examples/hybrid_DFT_band目录下有一个使用杂化泛函计算磷化镓的能带结构的例子。同样也可以使用DFT计算该磷化镓结构的能带,只是传统的DFT泛函会低估带隙。为了计算能带,首先得获得晶体的第一布里渊区内的一条或几条高对称点路径。在有POSCAR的目录下运行vaspkit,输入3选择功能K-Path for Band-Structure,在下一个界面输入3选择Bulk Structure,你会得到以下信息:
+-------------------------- Warm Tips --------------------------+
See An Example in vaspkit/examples/seek_kpath.
This Feature Is Experimental & Check Your System using SeeK-Path.
For More details See [www.materialscloud.org/work/tools/seekpath].
+---------------------------------------------------------------+
-->> (1) Reading Structural Parameters from POSCAR File...
+-------------------------- Summary ----------------------------+
Prototype: AB
Total Atoms in Input Cell: 2
Lattice Constants in Input Cell: 3.854 3.854 3.854
Lattice Angles in Input Cell: 60.000 60.000 60.000
Total Atoms in Primitive Cell: 2
Lattice Constants in Primitive Cell: 3.854 3.854 3.854
Lattice Angles in Primitive Cell: 60.000 60.000 60.000
Crystal System: Cubic
Crystal Class: -43m
Bravais Lattice: cF
Extended Bravais Lattice: cF2
Space Group: 216
Point Group: 31 [ Td ]
International: F-43m
Symmetry Operations: 24
Suggested K-Path: (shown in the next line)
[ GAMMA-X-U|K-GAMMA-L-W-X ]
+---------------------------------------------------------------+
-->> (2) Written PRIMCELL.vasp file.
-->> (3) Written KPATH.in File for Band-Structure Calculation.
-->> (4) Written HIGH_SYMMETRY_POINTS File for Reference.
-->> (5) Written POTCAR File with the Recommended Potential!
+---------------------------------------------------------------+
| * DISCLAIMER * |
| CHECK Your Results for Consistency if Necessary |
| Bug Reports and Suggestions for Improvements Are Welcome |
| Citation of VASPKIT Is Not Mandatory BUT Would Be Appreciated |
| (^.^) GOOD LUCK (^.^) |
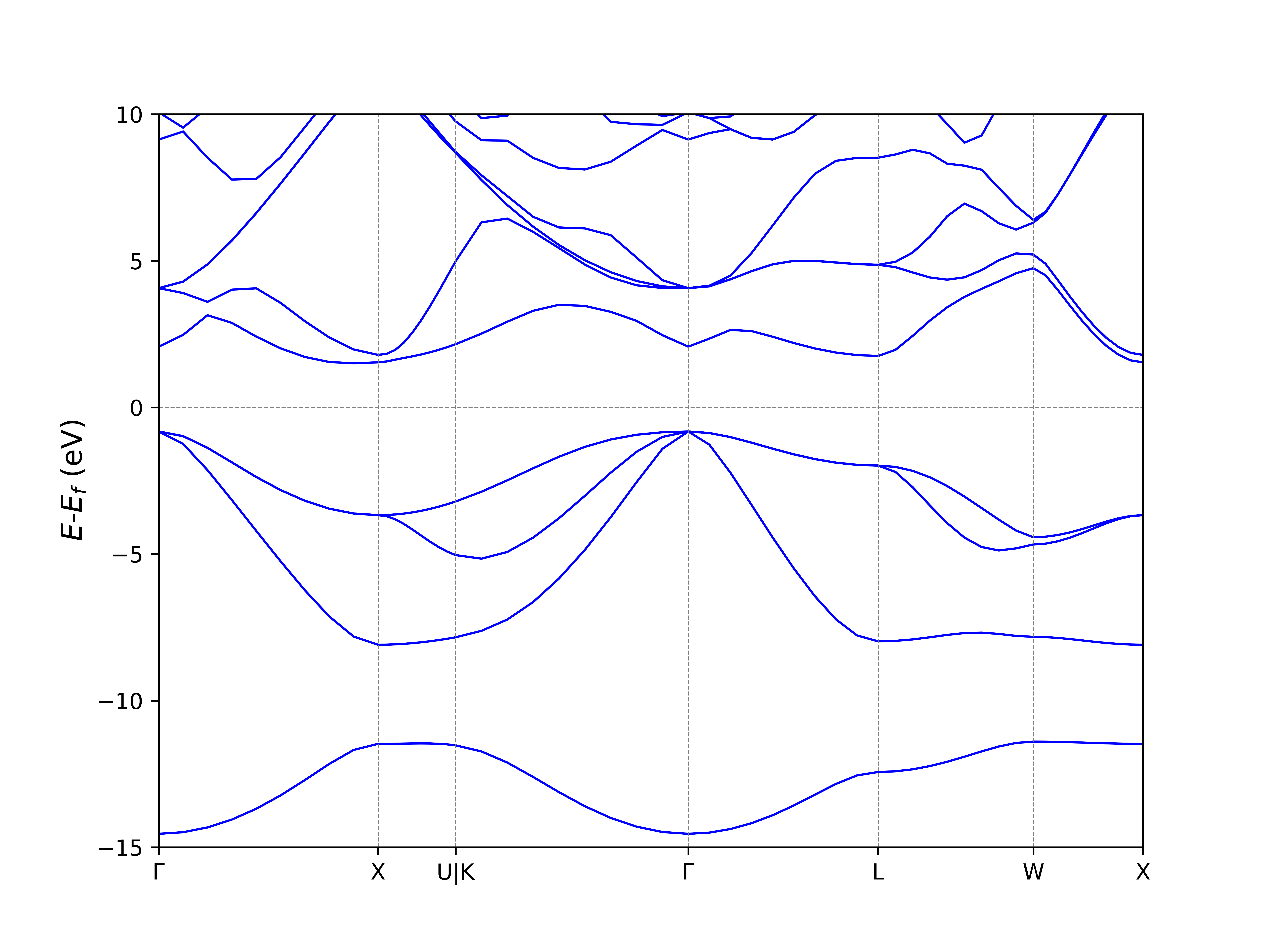
+---------------------------------------------------------------+vaspkit会分析晶体的对称性并得到两条建议的能带路径[ GAMMA-X-U|K-GAMMA-L-W-X ],同时生成了晶体的单胞结构PRIMCELL.vasp,并生成了KPATH.in用于能带结构计算。KPATH.in的第二行数字表示了每小段路径中插值的K点的数目,如果默认的数值都算不动的话,可以考虑将其设小。能带路径只针对于primitive cell,因此需要执行下面的命令,用生成的primitive cell作为计算的结构文件。务必执行以下操作,除非你知道你在做什么否则可能会得到错误的结果。
cp -f PRIMCELL.vasp POSCAR执行优化结构
对POSCAR(primitive cell)结构优化计算,得到平衡晶格常数。下图展示的是磷化镓的primitive cell和其第一布里渊区的高对称点位置,由SeeK-path网站生成。

PBE静态计算和计算能带
PBE泛函计算能带分为两步,第一步使用普通K点网格(功能102)进行自洽计算 ,启动vaspkit,输入1选择VASP Input Files Generator,再选择108选择Successive Procedure to Generate VASP Files and Check功能,输入ST,生成静态自洽的INCAR,并按照提示生成自洽用的K点。本例中ISMEAR=0,即Gaussian Smearing方法,如果是金属体系可以选择换成ISMEAR=1。接着调用VASP计算。第二步:使用KPATH.in里的高对称点信息作为新的KPOINTS,然后读入电荷CHGCAR进行能带非自洽计算,即:
cp -f KPATH.in KPOINTS 也就是读入上一步生成的CHGCAR(令INCAR=11)并保持不变,调用VASP计算。
待第二步完成后,通过功能21从能量本征值文件中读入能带结构。值得一提的是,费米能级应该以自洽计算的为准,因此如果想获得准确的费米能级,可以在自洽计算文件夹中执行提取态密度操作,vaspkit会同时生成FERMI_ENERGY文件,把这个文件重命名成FERMI_ENERGY.in并拷贝能带计算文件夹中,然后再次执行能带数据提取,如果FERMI_ENERGY.in文件存在,vaspkit则会从FERMI_ENERGY.in读取费米能级。如果想手动平移费米能级到价带顶,可在FERMI_ENERGY.in文件第二行设置价带顶能级大小保存后再次运行vaspkit即可。
如果您使用VASPKIT,请记得引用哦!
V. Wang, N. Xu, J.-C. Liu, G. Tang, W.-T. Geng, VASPKIT: A User-Friendly Interface Facilitating High-Throughput Computing and Analysis Using VASP Code, Computer Physics Communications 267, 108033, (2021), https://doi.org/10.1016/j.cpc.2021.108033
欢迎关注VASPKIT公众号。

「感觉有帮助?一键投喂 牛奶/咖啡/冰阔乐!」
 VASPKIT
VASPKIT
(๑>ڡ<)☆哇~太棒了!
使用微信扫描二维码完成支付

怎么在静态自洽文件夹路提取态密度